Reporting adverse drug reactions is critical for the pharmacovigilance system to work properly. The global and individual supervision over the safety of pharmacotherapy in various countries is an important element of healthcare systems worldwide.
It was agreed across the European Union that an adverse drug reaction is every unfavourable and unintended effect of a medicinal product. In some hospitals, though, the definition of an adr has been narrowed by excluding consequences of using drugs before hospital admission or in non-therapeutic doses.
Already in previous audits in 2016-2019, NIK established that adrs were not reported to relevant institutions and to hospital pharmacies or were reported with significant delays.
In the course of this audit on the adr monitoring, it was found that none of the audited hospitals made sure the system of detecting, reporting and analysing adverse drug reactions worked properly. In as much as 30% of the audited hospitals, the medical staff did not confirm any adrs and in 20% of hospitals the number of adrs confirmed by medical staff ranged from 2 to 8 cases.
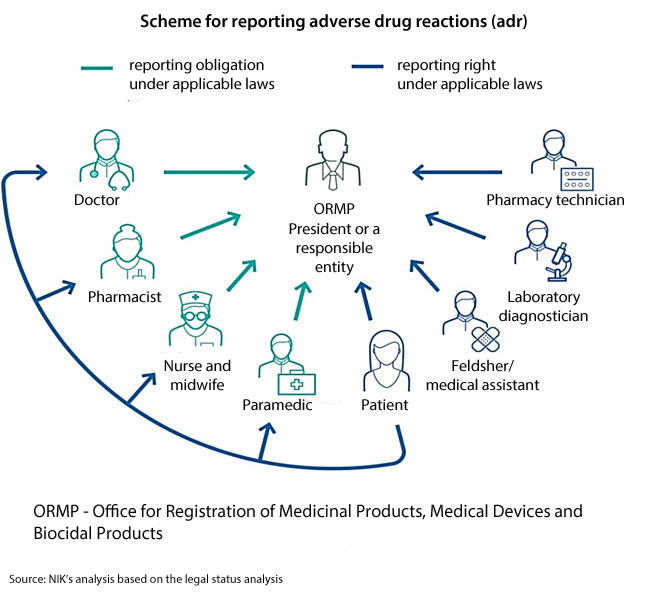
The adverse drug reactions, including vaccine adverse events, should be reported to a relevant institution or a responsible entity. Both the Office for Registration of Medicinal Products, Medical Devices and Biocidal Products (or ORMP) and responsible entities are obliged to make cause and effect analyses of the reports and pass them on to external databases. In Poland there are regional adr monitoring centres, operating as part of higher education schools in Cracow, Poznań, Wrocław, Szczecin and Gdańsk. Although they are independent of the ORMP, and formally are not part of the adr reporting system, they gather and analyse data about adverse drug reactions and also conduct information and training activity for healthcare workers in terms of adr reporting.
In case medical staff did not consider some undesired drug effects as adverse drug reactions, although the medical documentation indicated they should have, the monitoring authorities were not aware of the adr occurrence. That weakened the system.
The issue of too low percentage of identified and reported adrs against their real number was addressed by representatives of doctor communities and experts in pharmacotherapy safety, also during the panel organised at NIK headquarters in February 2020. The meeting was attended by representatives of the Office for Registration of Medicinal Products, Medical Devices and Biocidal Products, the National Health Fund, the Commissioner for Patient’s Rights, scientific communities, professional association of pharmacists and associations whose core activity is related to drug use safety. All the debate participants along with NIK disapproved of the fact that adverse drug reactions were not reported to the Office for Registration of Medicinal Products, Medical Devices and Biocidal Products.
The adverse drug reactions are usually reported by doctors, whereas nurses and paramedics are not willing to do so. The audit findings and the survey results of 840 respondents also reveal that persons performing medical professions happen to wrongly interpret the definition of an adverse drug reaction, do not know this definition or fail to apply it.
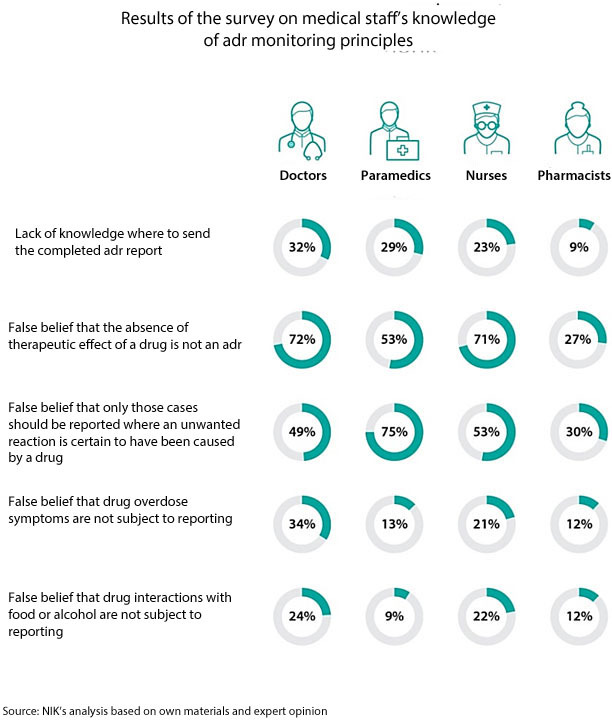
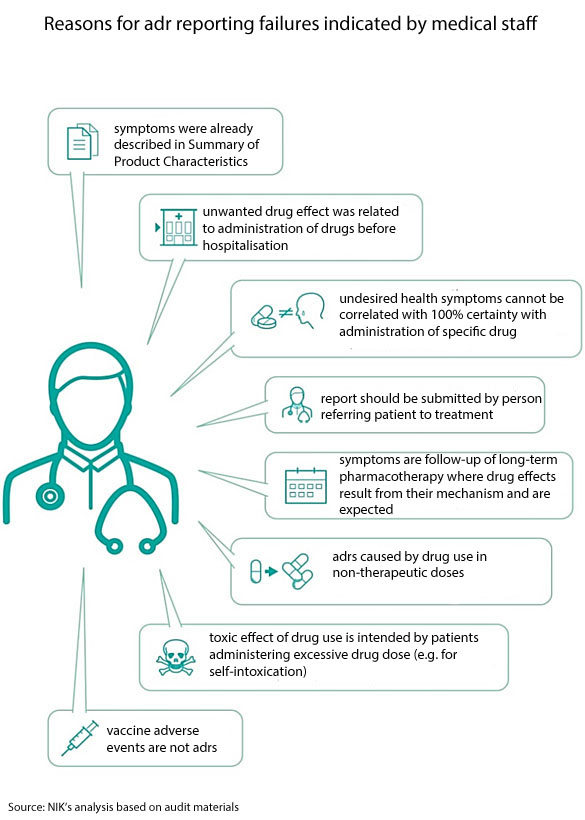
In over 300 cases (out of 1190 cases analysed by NIK), the medical staff did not consider negative drug effects as adrs, although the conditions for such qualification were met. As much as 85% of those cases, in line with the statutory definition, should be considered as serious adverse drug reactions as they required hospitalisation. According to NIK findings, the number of reported adrs in the audited entities should be twice as many as occurred in reality. As a consequence, a big part of unwanted drug effects are not reported or are wrongly classified (”non-serious” instead of ”serious” reactions).
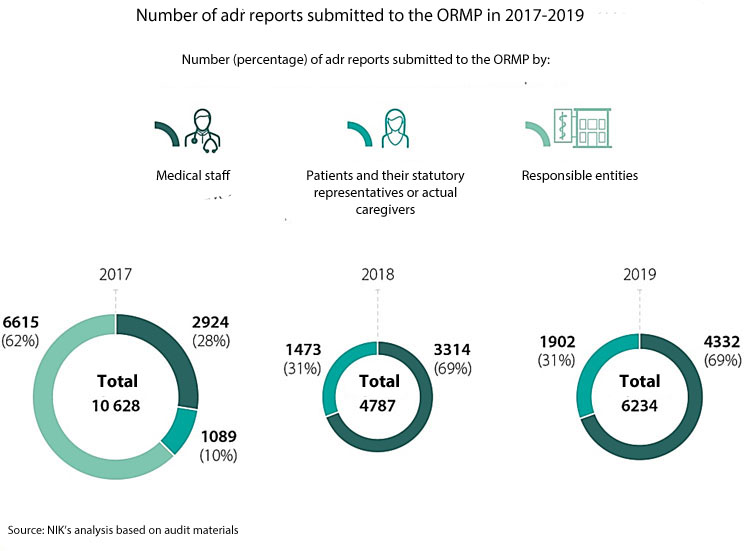
Nearly half of the adrs confirmed by medical staff in the audited hospitals were not reported to the Office or a responsible entity or were reported with delays exceeding the statutory deadline of 15 days.
None of the audited hospitals provided conditions for the medical staff to meet their obligations in terms of reporting adrs. There is no supervision in those hospitals over the way adrs are identified and reported. Besides, the scale of information and education campaigns was too small. They were carried out only in ¼ of hospitals, in a limited scope and for a small group of employees. The involvement of hospital pharmacies in monitoring adrs is not too high and doctors do not provide those facilities with adr data.
Only medical staff are obliged to report adverse drug reactions. Medical facilities are not obliged to pass on the reports further which makes enforcement of that obligation in hospitals much more difficult.
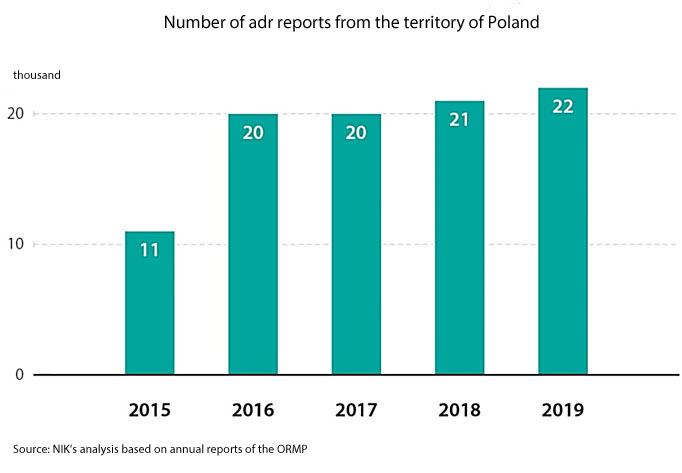
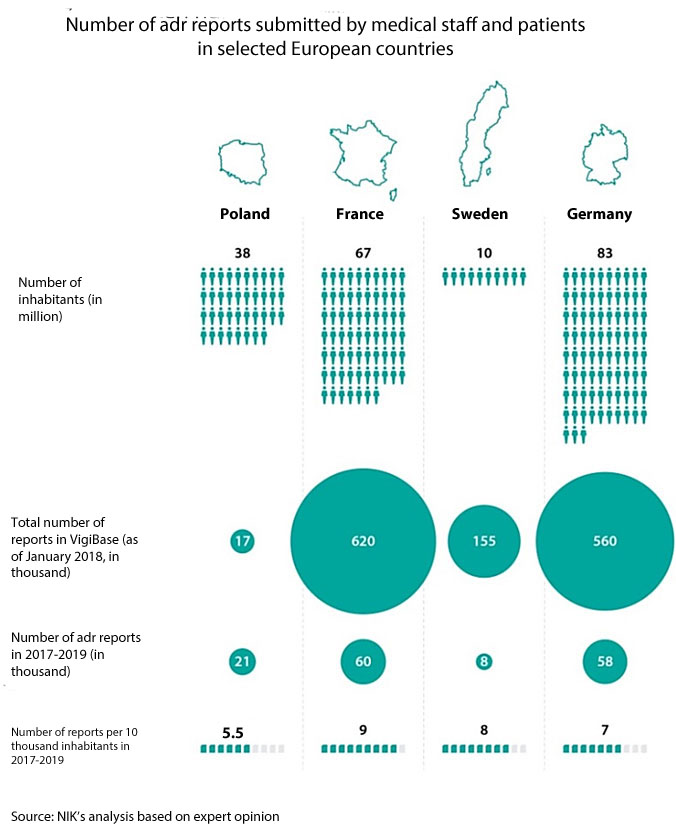
The data obtained by an expert appointed in the course of the audit reveals that doctors in Poland submit over 50% fewer adr reports than could be expected. Considering the number of doctors, people and medicinal products it is expected that about 45 thousand adrs should be reported per year. In reality it is from 19996 to 21696 reports per year (data of 2017-2019).
The Office for Registration of Medicinal Products, Medical Devices and Biocidal Products properly executed some tasks related to pharmacovigilance. The ORMP posted relevant information on the safety of using medicinal products, took measures to activate persons obliged or entitled to report adrs. The monitoring of adrs did not work properly which could be noticed above all in long-term delays of the ORMP in conducting cause and effect analyses and in passing on the reports to external databases. The ORMP reduced delays in analysing submitted reports initially to four years. Though, the number of reports passed on with delays to external databases increased in the audited period from nearly 7 thousand to almost 19 thousand.
Thus, very important data on noxiousness of certain drugs did not reach (or reached with delays) the institutions responsible for monitoring the safety of drugs on the European and global scale. As a consequence, drug producers and medical staff did not receive that data on time, either.
NIK has pointed out that delays in submitting data were related among others to staff shortages in the Office. The ORMP President announced subsequent recruitments but they did not bring about the expected effects. He also informed the Minister of Health about the need to increase headcount in the Office due to new obligations under the applicable laws. NIK has stressed that the effectiveness of the adr monitoring system is determined by the organisational strengthening of the ORMP. It is particularly important as the issues with executing statutory tasks related to the adr monitoring may deepen along with growing awareness of persons obliged and entitled to reports adrs.
Recommendations
General recommendations to the Minister of Health
Following the audit findings, NIK has requested the Minister of Health to:
- conduct a detailed analysis of the ORMP’s financial needs essential for proper discharge of obligations related to the analysis and submittal of adr reports,
- be more involved in the process of educating future doctors and other medical staff adr-related issues and constantly raise qualifications in this area.
De lege ferenda proposals to the Minister of Health
Legal solutions related to the adr monitoring did not provide conditions for proper information flow about adr occurrence. Therefore, the Supreme Audit Office has also addressed the following proposal to the Minister of Health:
- to consider changing the law by introducing the obligation to record in the medical documentation an adr occurrence including its category;
Adequate pharmacovigilance requires that an adr occurrence should be documented and recorded by medical staff.
The said change will make it possible to:
- supervise the discharge of adr reporting obligations, under special laws, by medical staff;
- specify if the given treatment process may be linked to an adr occurrence;
- optimise decisions made by persons performing medical professions who will be able to consider, on a comprehensive basis, potential benefits and threats of proposed therapeutic solutions.
- take legislative measures to introduce the term ”monitoring adverse drug reactions” in the effective laws.
Interpretation of the involvement in the adr monitoring referred to in the Polish law varies among persons responsible for the operation of hospitals. In some cases this pharmaceutical service amounts to limiting the role of hospital pharmacy managers to recording adverse drug reactions and submitting them to the ORMP or a responsible entity.
System recommendations to hospital directors
In view of the findings of the audit conducted in hospitals NIK has recommended as follows:
- make sure the adr monitoring is effectively supervised, including identification and classification of adverse drug reactions by medical staff and their timely reporting, as well as proper discharge of obligations under internal adr procedures;
- make sure the adrs in hospital patients are reported to the ORMP President or a responsible entity and hospital pharmacies in line with effective deadlines; then the adrs should be recorded;
- make sure hospital pharmacies are involved in the adr monitoring;
- take or intensify information and education measures addressed to medical staff; covering the adr monitoring and reporting issues.
